ISO 13485 – Medical Devices – Quality Management System

ISO 13485 – Medical devices — Quality management systems — Requirements for regulatory purposes specifies requirements for a quality management system where an organization needs to demonstrate its ability to provide medical devices and related services that consistently meet customer requirements and regulatory requirements applicable to medical devices and related services.
The primary objective of ISO 13485 is to facilitate harmonized medical device regulatory requirements for quality management systems. As a result, it includes some particular requirements for medical devices and excludes some of the requirements of ISO 9001 that are not appropriate as regulatory requirements. Because of these exclusions, organizations whose quality management systems conform to this International Standard cannot claim conformity to ISO 9001 unless their quality management systems conform to all the requirements of ISO 9001.
All requirements of ISO 13485 are specific to organizations providing medical devices, regardless of the type or size of the organization.
If regulatory requirements permit exclusions of design and development controls, this can be used as a justification for their exclusion from the quality management system. These regulations can provide alternative arrangements that are to be addressed in the quality management system. It is the responsibility of the organization to ensure that claims of conformity with ISO 13485 reflect exclusion of design and development controls.
1) certain ISO-9001 requirements and
2) newly defined requirements catering specifically to the medical device industry.
As such, ISO-13485 differs from ISO-9001 in certain ways, modifying or even excluding some of the latter’s requirements. For instance, the ISO-13485 excludes the ISO-9001’s requirements related to continual improvement because most medical device regulations require organizations to maintain their quality management systems, and not to improve on them. Thus, while ISO-9001emphasizes the importance of improving quality systems, ISO-13485 emphasizes the importance of maintaining them. ISO-9001 customer satisfaction requirements were also excluded because some of the committee members who worked on ISO-13485 found them to be too subjective
Some key points adopted by the ISO-13485 include:
1) focus on meeting regulatory requirements;
2) focus on meeting customer requirements;
3) use of a ‘process’ approach;
4) maintenance of the effectiveness of quality management systems; and
5) maintenance of procedural documentation.
As mentioned, the ISO-13485 has special requirements that are not covered by ISO-9001:2015. These special requirements include both documentation and system/process requirements that cater to the medical device industry.
Special system/process requirements of the ISO-13485 include:
1) risk management systems;
2) clinical evaluations and trials;
3) product cleanliness and contamination controls;
4) requirements for implantable devices;
5) proper communication of advisory notices; and
6) additional research and development requirements.
Aside from regulation-required documents, additional documentations required by ISO-13485 include those pertaining to:
1) responsibilities and authorities;
2) training procedures;
3) health, cleanliness, and clothing;
4) environmental conditions;
5) control of contaminated products;
6) risk management;
7) customer requirements;
8) design and development;
9) purchasing control, including purchase traceability and verification;
10) reference materials;
11) labeling and packaging;
12) installation and verification;
13) sterilization process validation;
14) preservation of product (including shelf life); and
15) measurement and monitoring.
Difference between ISO 9001 and ISO 13485
Some of the specific requirements of ISO 13485 are discussed below. The focus is on the differences between ISO 9001 and ISO 13485. Please check the standard for the exact language and requirements. The standard emphasizes the exact differences within the text of the publication.
In ISO 13485 there is a change in emphasis from “improving” the effectiveness of the quality system to “maintaining” the effectiveness and on meeting customer and regulatory requirements versus increasing customer satisfaction. There is more documentation required in ISO 13485 also.
– The additional required documentation includes:
– Those documents required by regulation
– Responsibility and authority
– Training procedure, if required
– Maintenance requirements
– Health, cleanliness, clothing
– Environmental conditions
– Control of contaminated product
– Risk management
– Customer requirements
– Design and development
– Purchasing process including traceability records and records of verification
– Reference materials and reference measurement procedures
– Labeling and packaging
– Installation and verification
– Sterilization process validation
– Identification and validation
– Preserving product including shelf life
– Monitoring, measurement and measuring devices
– Sterilization records, servicing records, batch records, validation
Differences in design and development activities:
– Determine design transfer activities
– Document design planning outputs
– Include risk management in input
– Approve inputs
– Document design outputs
– Include specialist as needed in design review
– Complete validation before delivery
– Include clinical trial as required
Many people in the medical device industry do not know much more about quality systems than that they are required. This article provides an overview of medical device quality systems and then describes generally the requirements of the ISO 13485 international standard for medical devices quality management systems (QMS). Medical devices can be simple or complex, but all of these can benefit from being designed and manufactured under ISO 13485 which is the most widely used medical device QMS standard. It is required in Europe, Canada and many other countries for most devices. In the US the FDA Quality System Regulation (QS Reg.), also known as the cGMP, is required. Although the QS Reg. is structured very differently than ISO 13485, they have no conflicting requirement
ISO 13485 is a regulatory standard whose focus is meeting customer requirements, including regulatory requirements, and maintaining the effectiveness of the QMS. This differs from ISO 9001:2015 which focuses on customer satisfaction and continual improvement. Whereas both customer satisfaction and continual improvement are as important to medical device manufacturers as to any other business today, these things are hard to measure and tend to be somewhat subjective. So when it came time to adapt ISO 9001:2015 to the medical device industry, these potentially subjective requirements were changed to meeting customer requirements and maintaining the effectiveness of the QMS, which are more easily measureable. The other major difference from ISO 9001, which is also consistent with the fact that this is a regulatory standard, is that there are more requirements for documented procedures. In ISO 13485, meeting requirements includes meeting regulatory requirements. So for devices that will be used in the US, to be compliant with ISO 13485, manufacturers must also meet the QS Reg. As a regulation the QS Reg. is often more specific than ISO 13485, particularly in the areas of complaint handling, labeling control, and documentation. ISO 13485 is structured the same way as ISO 9001:2015, and is in fact about 90 % the same as this general standard for quality management systems. The reason for the differences between ISO 13485, ISO 9001 and the FDA QS reg., can be understood by looking at the differences in their objectives as given below.
Objective of ISO 13485
– To set out requirements for a QMS that is capable of consistently meeting customer requirements, including regulatory requirements.
Objective of ISO 9001:2015
– To set out requirements for a voluntary, generic QMS that is capable of meeting customer & regulatory requirements, and enhancing customer satisfaction through process including continual improvement.
Objective of the FDA Quality System Regulation
– To set out requirements for a QMS that is capable of consistently providing safe and effective medical devices.
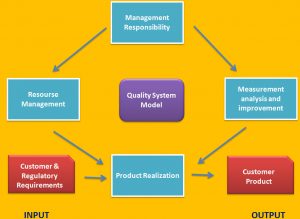
A good QMS, if integrated into the goals and management of a company, provides a way to reduce variation. Reducing variation can provide financial benefits for the company, such as reduced scrap and general process efficiencies. So in addition to being a regulatory requirement, a well-functioning QMS makes good sense from a business and financial perspective. ISO 13485 follows the process approach introduced in ISO 9001:2015. The process approach treats the QMS as a set of interrelated processes covering not only the manufacture of a product or provision of a service, but also management processes and support processes. A “process” is something that transforms a collection of inputs into outputs. Inputs consist of everything needed to accomplish this transformation. For manufacturing a device these this might included such things as raw materials, manufacturing supplies, work benches, cleaning materials, tools, and equipment, the building, people, written instructions, assembly drawings, comparison samples, and workmanship standards. The output of the process, that is the transformation of these inputs, produces the finished part, records about what was done by who, and information about how the transformation was accomplished, such as time to complete or production yield. Unwanted outputs might include scrap parts and wasted material. For non-manufacturing processes, for example Document Control, inputs might include Document Control procedure, change request, people, equipment (copy machine, computer, scanner), document control center, and the outputs would included controlled documents, controlled copies, and process statistics. As you can see from even just these two examples, the output of one process, i.e. Document control, is the input to other processes, such as manufacturing. Below figure gives a diagram of how the ISO 13485 standard is organized.
ISO 13485 Section 4 gives the general requirements. These include identifying specific processes and how they interact, and responsibility for processes that are outsourced. A quality manual, quality policy and objectives and the requirements for control of documents and records and for outlining the company’s document structure are given in Section 4. Document control includes review and approval of documents before use, control of changes, and making sure that current versions of controlled documents are available where needed for use. Requirements for control of records include maintaining their integrity and establishing procedures for how long documents and records are maintained.
The management of a company must take an active part in the establishment and maintenance of an ISO 13485 QMS. Section 5 requires management involvement at the level of the person who makes policy and financial decisions. This is usually either the CEO or the chief of operations. Establishing the quality policy and objectives, support and oversight of the QMS and provision of resources are the direct responsibility of upper management. In addition, top management appoints a Management Representative, usually the most senior quality manager, who has the day-to-day responsibility for the functioning of the QMS. Upper management’s commitment must also include quality planning, and making sure that the quality policy is understood at every level of the organization.
There are specific requirements for the periodic management review of the QMS. This specifies the minimum of what must be covered in these reviews, as well as the output requirements. This is one of the most important processes for a QMS, and also adds value to the company by providing a structured framework managing for quality and productivity.
Section 6 contains requirements for provision of resources. Management must assure adequate facilities including, space, tools, and equipment, including computer systems. The building environment must fit the devices being made, including where necessary, such environments as clean rooms. Buildings, tools and equipment must be maintained in order to produce devices meeting all their requirements. The QMS must have as process to insure that all required maintenance activities are preformed.
Human resources are essential to quality medical devices. Therefore the provision of and adequate number of people that are competent, capable, and aware of their job responsibilities is key. It is not sufficient to train personnel and keep good training records, although that is important. Management must first define job requirements, often in the quality manual and positions descriptions. The QMS must then document that employees meet these requirements, or have had training to fill in any gaps. Ongoing employee awareness of QMS requirements, particularly related to documents and recordkeeping is the responsibility of management. Employees must also have awareness of their job responsibilities, including their responsibilities for product quality. They must know the consequences to the product or to the people using the product, if they fail to do their job properly.
The portion of the standard that most effects what people in the company do on a day-to-day basis is section 7, with the unusual name of “Product Realization.” This covers much more than manufacturing. It does in fact cover everything that is required to realize a product, from customer requirements to creating (designing and manufacturing), installing and supporting a medical device.
Planning is an essential part of a functioning QMS, and in planning for product realization the company is required to establish processes for all phases of product realization, from how they obtain customer requirements, design products, purchase supplies and materials, make, install and service a device. There is risk associated with everything that we do, but in making medical devices these can include the risk to a person’s life. Therefore ISO 13485 requires that “The organization shall establish documented requirements for risk management throughout product realization.” Risk management includes the following:
– Risk Assessment – Identifying risks
– Risk Analysis – looking at severity and probability of all hazardous situations
– Risk Reduction – reduction, mitigation (labeling), elimination of risk as much as possible or practical
Risk management applies to processes, including all QMS processes. However, most importantly it applies to device design, manufacturing and support processes. This is such an important process that ISO 13485 requires that risk management be done according ISO 14971, the international standard for medical device risk management.
Planning for product realization begins with establishing processes for handling customer requirements, and how to communicate with the customer throughout the lifecycle of the device. Requirements may be as simple as processing orders from the company’s catalog, to as complex as requirements to design a complex device from a general concept. Communication includes back and forth communication with the customer on requirements changes, and way of collecting customer feedback on all aspects of the device and the manufacturer’s business processes.
If a company does product or process design, they must follow the requirements for design controls given in ISO 13485. When governments and regulatory agencies looked at reported adverse events of medical devices, they found that as often as not the problems were caused by poor design. So having a controlled design process that includes risk management, verification, validation and controlled transfer of a design to manufacturing can reduce the potential for adverse effects. A product development process following the design control requirements begins with establishing design requirements, and goes through validation and transfer to manufacturing, as outlined in below.
– Design and development planning
– Design Input
– Risk Management
– Design Output
– Design Review
– Design Verification
– Design Validation
– Design Transfer
– Design Changes
– Design History File (DHF)
Once there is a device design with established manufacturing processes, it is important to make sure that the materials going into and used in making the device are correct. ISO 13485 purchasing requirements cover purchasing from qualified suppliers, according to pre-established specifications, and assuring that purchased product meets those specifications.
Manufacturing or production processes must be controlled to assure that the manufactured device meets all of its specifications. This includes not only controlling the production processes, but control of how material and devices are identified, stored and used. Documented processes must cover receiving, warehouse, production, testing, shipping, installation and servicing. Some of these processes cannot or cannot economically be fully tested to assure that all product specifications are met. Processes that cannot or will not be fully verified must be validated to assure that they always meet specifications, and once validated must be controlled and performed by trained personnel.
One of the ways to insure that a product meets its specifications involves the use of monitoring and measuring equipment. This equipment must be controlled to assure that it gives accurate results. A calibration and preventive maintenance program is essential to this control.
The last section of ISO 13485 is the one that provides the feedback and other information that allows management to maintain the effectiveness of the QMS and includes:
– Feedback including Customer Complaints and handling adverse events
– Internal audit
– Monitoring and measurement of processes
– Monitoring and measurement of product including nonconforming product
– Analysis of data
– Corrective and preventive action
A corrective action is one that fixes the root cause of a problem that has happened. This is often confused with fixing a problem that exists. Just fixing a problem is not sufficient. A root cause analysis that can be as simple as asking “WHY” five times, is not only essential to a corrective action system, but to the effectiveness of the entire QMS. Preventive action, on the other hand, is a system that if used successfully will provide one of the largest financial benefits of the QMS. Preventive actions are taken to prevent nonconformities by fixing things that might go wrong.
There are key steps that every company implementing a QMS will need to consider:
Purchase the Standard
Before you can begin preparing for your application, you will require a copy of the standard. You should read this and make yourself familiar with it.
Review support literature and software
There are a wide range of quality publications and software tools designed to help you understand, implement and become registered to a quality management system.
Assemble a team and agree on your strategy
You should begin the entire implementation process by preparing your organizational strategy with top management. Responsibility for a QMS lies with Senior Management, therefore it is vital that Senior Management is involved from the beginning of the process.
Consider Training
Whether you are the Quality Manager seeking to implement a quality management system or a Senior Manager who would like to increase your general awareness of ISO 14971, Risk Management etc there are a range of workshops, seminars and training courses available.
Review Consultancy Options
The consultancy is the 3rd party like NUCLEUS, You can receive advice from independent consultants on how best to implement your quality management system. They will have the experience in implementing a QMS and can ensure you avoid costly mistakes.
Choose a registrar
The registrar is the 3rd party, like (Check with International Accreditation Forum for the accredited certification body) who come and assess the effectiveness of your quality management system, and issue a certificate if it meets the requirements of the standard. Choosing a registrar can be a complex issue as there are so many operating in the market. Factors to consider include industry experience, geographic coverage, price and service level offered. The key is to find the registrar who can best meet your requirements. A great place to start is by contacting us.
Develop a Quality manual
A Quality manual is a high level document that outlines your intention to operate in a quality manner. It outlines why you are in business, what your intentions are, how you are applying the standard and how your business operates.
Develop support documentation
This is typically a procedures manual that supports the Quality manual. Quite simply, it outlines what you do to complete a task. It describes who does what, in what order and to what standard.
Implement your Quality Management System
The key to implementation is communication and training. During the implementation phase everyone operates to the procedures and collects records that demonstrate you are doing what you say you are doing.
Conduct Internal Audit and Management Review
At planned intervals you need to conduct an internal audit and a management review to verify the established QMS is confirming the applicable requirements and it is effective, suitable and adequate.
Gain registration
You should arrange your initial assessment with your registrar. At this point the registrar will review your QMS and determine whether you should be recommended for registration.
Continual assessment
Once you have received registration and been awarded your certificate, you can begin to advertise your success and promote your business. To maintain your registration, all you need to do is continue to use your quality system. This will be periodically checked by your registrar to ensure that your Quality System continues to meet the requirements of the standard.
QMS Registration
Registration to ISO 13485 takes place when an accredited 3rd party visits an organization, assesses the management system and issues a certificate to show that the organization abides by to the principles set out in ISO 13485.
Your Quality Management Systems certificate is a sign of acceptability which saves you having to prove your quality standards to discriminating customers. ISO 13485 speaks an international language. Once you are certified means, the certificate is valid for 3 years and it is subject to the completion of the annual surveillance audit.
Why do I need registration?
Gaining registration to ISO 13485 through various certification bodies will help your organization flourish. Whether you are looking to operate internationally or to expand locally to accommodate new business, ISO 13485 will help you demonstrate to customers that you have a commitment to quality.
The regular assessment process will ensure you continually use, monitor and improve your processes.
Registration can improve overall performance, remove uncertainty and widen market opportunities.